在人类肠道菌群中检测到生态进化反馈
来自斯坦福大学的Benjamin H. Good 等人,最近在《自然通讯》上报道了人类肠道菌群的演化及其对群体构成的影响。他们表示,虽然菌群随时间会进化,但短期进化对菌群整体构成的影响尚不完全清楚。在这项研究中,他们使用基于参考的方法来识别菌群内的遗传修饰,并研究这些修改如何影响群体构成。为了探索这一点,研究人员分析了一个跨多个物种和宿主的遗传比较数据集。他们发现,进化事件并不是均匀分布在各个物种中,这表明进化可以改变菌群的生态组成。此外,置换和修改事件都显著影响了生态系统结构,表明菌群内部存在复杂的生态进化反馈。这意味着一个物种内的遗传变化可能导致其他物种丰度的相关性变化。这项研究强调了理解肠道微生物群短期进化的重要性,以便理解整个菌群的组成。研究发现表明,菌群的结构和功能可能受到它们本地进化历史的影响,这可能对个性化医疗和菌群工程学有所启示。因此,他们旨在调查菌群内遗传修饰所导致的群体构成变化,并探索这一复杂生态系统内的潜在生态进化反馈。
他们执行了一个宏基因组工作流程。宏基因组工作流程是一种用于分析和解释宏基因组数据的计算工具。它遵循逐步过程从复杂的微生物群落中提取有意义的信息。此管道涉及多个阶段,包括数据预处理、质量控制、组装、基因预测、功能注释和分类学分类。在数据预处理阶段,对宏基因组序列进行修剪和过滤,以移除低质量读序列和接头。然后,在组装步骤中,剩下的高质量读序列被组装成更长的连续序列,称为contigs。接下来,应用基因预测算法来识别contigs中的基因。然后,这些预测出的基因会通过比对现存的数据库进行功能性注释,即赋予它们推定的功能。此外,分类学分类阶段涉及将contigs或预测基因分配到特定的分类学群体,以确定微生物群落的组成。宏基因组管道提供了一种结构化的方法来处理和分析宏基因组数据,使研究人员能够洞察微生物群落在不同环境中的基因潜力和功能能力。它被证明是一个宝贵的工具,用于了解微生物在其生态位中的复杂相互作用和角色。
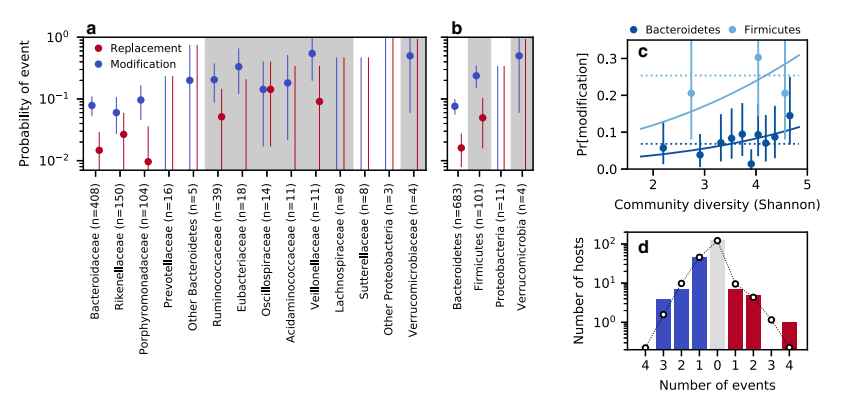
他们使用数据来识别物种内部随时间的置换和修改事件。他们专注于有着简单谱系结构的“准可相位”物种,可以确信地识别出占主导地位的谱系。研究人员计算了每个准可相位群体内,在不同时间点之间,从低频到高频(或相反)转变的单核苷酸变异(SNVs)数量。他们还考虑了测序噪声,以估计假阳性率。基于这些计数,他们将每个群体分类为经历了一个置换事件、一个修改事件或没有遗传变化。由于可能的样品交换,两个受试者被排除在外。分析揭示了不同物种和宿主共计18个置换事件和98个修改事件。为了避免重叠的时间间隔,数据进一步去重,得到减少数据集中的16个置换事件和78个修改事件。这些发现被用于后续分析。
研究检查了物种内部进化的速率如何受到本地群体组成的影响。研究人员发现,菌株置换和进化修改事件在分类群中的分布并不均匀。一个总检测试验显示,在尤其是在家族或门水平上,横跨物种有明显的变异富集。拟杆菌门和厚壁菌门之间的差异在推动这些模式方面起着主要作用,其中厚壁菌门的置换和修改事件发生率更高。研究还调查了进化速率与群体多样性之间的关系,但仅发现了微弱的正相关。群体多样性并没有强烈地限制健康人类肠道菌群进化的速率。研究表明,菌群内的遗传变化受到全局社群变量之外因素的影响,并为理解微生物群落内物种进化的动态提供了见解。
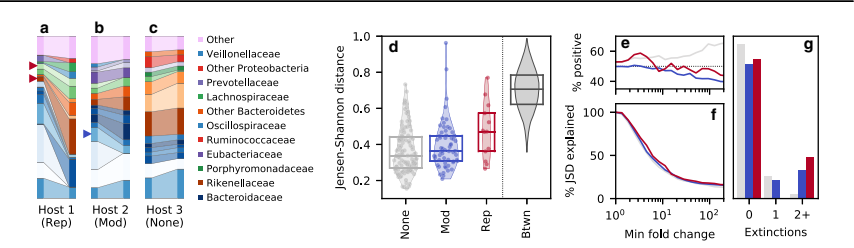
他们在一个名为拟杆菌属粪拟杆菌的物种中检测到一个进化修改事件。在这次事件中,拟杆菌属粪拟杆菌的相对丰度略有下降。然而,他们也观察到在同一时间间隔内有两个其他物种灭绝。这些灭绝物种中的一种,拟杆菌属马赛拟杆菌属于同一属,而另一个灭绝物种,巴内斯菌属肠杆菌属于不同的细菌家族。为了更全面地检查这些物种之间的关系,作者进行了系统分析。他们计算了一个被称为J SD的演化修改事件量,该量由作为焦点物种之一的同一家族的物种贡献。当考察J SD的部分时,作者观察到与拟杆菌属粪拟杆菌同一家族的物种发挥了重要作用。这个分析为这些物种之间的关系提供了一个定量度量。总体而言,此部分突出了关于拟杆菌属粪拟杆菌中进化修改事件的作者发现。通过比较相对丰度的变化和相关物种的灭绝情况,作者得以洞察这些物种如何相连,以及它们如何贡献于观察到的进化模式。系统分析让人们能够更深入地理解微生物群落内的关系。
研究人员进行了分析,调查人类肠道菌群短期进化与其生态结构变化之间的相关性。他们提出了两种可能的因果情景:进化驱动反馈,其中遗传变化改变了物种之间的生态相互作用;以及生态驱动反馈,其中环境干扰导致分类群的变化,并为宿主内部进化创造了新的机会。为了更好地理解这些情境,研究人员用了一个简单的资源竞争数学模型。在这个模型中,共存的物种为环境持续供应的可替代资源进行竞争。模型显示,在生态平衡的饱和社群中,新突变的选择压力与外部环境和居民物种的丰度无关。这表明生态驱动反馈被社群的集体响应有效地抑制了。研究人员还推导出突变入侵健康度与其引发的生态干扰之间的关系。这种关系显示,突变可能导致一系列物种中的正面和负面丰度变化,与它们的系统发育或表型相似性无关。这些发现支持了简单的进化驱动反馈可以解释在肠道菌群中观察到的遗传和生态变化之间的相关性的观点。
然而,研究人员也承认他们的分析有局限性。理论结果仅适用于饱和社群中小效应突变的最简单情况。需要进一步的理论工作,以将这些结果扩展到非饱和社群,并纳入其他重要因素,如交叉喂养和肠道内的空间结构。此外,需要更多关于资源摄取表型的遗传结构信息,以确定相应变化多容易由居民菌株中的突变产生。
总体而言,研究提供了对观察到的人类肠道菌群中的遗传和生态变化之间相关性背后潜在机制的见解。需要进一步的实验和理论工作,才能完全理解这些机制,并描绘出由突变导致的可达生态干扰。